Anlage 16
QUANTITATIVE BESTIMMUNG DES AMMONIAKS
- 1. ANWENDUNGSBEREICH
Die Methode beschreibt die Bestimmung von freiem Ammoniak in kosmetischen Erzeugnissen.
- 2. DEFINITION
Der nach dieser Methode bestimmte Gehalt der Probe an Ammoniak wird ausgedrückt in Massenprozent NH3.
- 3. PRINZIP
Zu dem kosmetischen Erzeugnis wird in Methanol-Wasser-Medium eine Bariumchlorid-Lösung hinzugefügt. Dieses Vorgehen vermeidet die Miterfassung bestimmter Ammoniumsalze während der Wasserdampfdestillation (wie des Karbonats und des Hydrogenkarbonats, von Salzen von Fettsäuren usw.). Ammonium-acetat wird unter diesen Bedingungen miterfaßt.
Das Ammoniak wird durch Wasserdampfdestillation aus dem Filtrat oder aus der überstehenden Flüssigkeit entfernt und titrimetrisch oder potentiometrisch bestimmt.
- 4. REAGENZIEN
Alle Reagenzien müssen analysenrein sein.
- 4.1. Methanol.
- 4.2. 25%ige (m/v) Bariumchlorid-Dihydrat-Lösung.
- 4.3. 4%ige (m/v) Ortho-Borsäurelösung.
- 4.4. Schwefelsäure-Maßlösung, 0,5 N.
- 4.5. Flüssiges Antischaummittel.
- 4.6. Natriumhydroxid-Maßlösung, 0,5 N.
- 4.7. Indikator: Mischung von 5 ml einer Methylrotlösung (0,1 % in Ethanol) mit 2 ml einer Methylenblaulösung (0,1 % in H2O).
- 5. GERÄTE
- 5.1. Übliches Laborgerät.
- 5.2. Zentrifuge mit verschließbaren Zentrifugengläsern.
- 5.3. Wasserdampfdestillationsapparatur.
- 5.4. Potentiograph.
- 5.5. Glaselektrode und Diquecksilberdichlorid(Kalomel)-Referenzelektrode.
- 6. VERFAHREN
- 6.1. In einen 100-ml-Meßkolben wird eine Probenmenge, die maximal 150 mg NH3 entspricht, auf 1 mg genau eingewogen.
- 6.2. Zu der Probe werden hinzugefügt: 10 ml H2O, 10 ml Methanol (4.1), 10 ml Bariumchloridlösung (4.2). Bis zur 100-ml-Marke mit Methanol (4.1) auffüllen.
- 6.3. Homogenisieren und über Nacht im Kühlschrank (5 °C) aufbewahren.
- 6.4. Die noch kalte Lösung wird dann filtriert oder in geschlossenen Gläsern zentrifugiert, bis eine klare Trennung erreicht ist (10 Min.).
- 6.5. 40 ml der klaren Lösung werden mit der Pipette in die Apparatur (5.3) eingefüllt und eventuell 0,5 ml Antischaummittel (4.5) hinzugegeben.
- 6.6. Anschließend wird destilliert. 200 ml des Destillats werden in einem 250-ml-Becherglas, das 10,0 ml H2SO4 – 0,5 N (4.4) und 0,1 ml des Indikators (4.7) enthält, aufgefangen.
- 6.7. Die überschüssige H2SO4 wird mit NaOH 0,5 N (4.6) rücktitriert.
- 6.8. Im Fall einer potentiometrischen Bestimmung werden 200 ml Destillat in einem 250-ml-Becherglas, das 25 ml Orthoborsäure (4.3) enthält, aufgefangen. Anschließend wird mit H2SO4 – 0,5 N (4.4) titriert.
- 7. AUSWERTUNG
- 7.1. Berechnung bei Rückbestimmung mit Indikator
Mit:
V1 (ml): Volumen der zugegebenen NaOH 0,5 N (4.6),
T1: Titer der NaOH 0,5 N (4.6),
T2: Titer der H2SO4 0,5 N (4.4),
m (mg): Probeneinwaage (6.1)
ergibt sich folgende Formel:
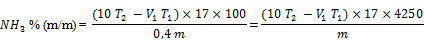
- 7.2. Berechnung bei direkter potentiometrischen Bestimmung
Mit:
V2 (ml): Volumen H2SO4 0,5 N (4.4),
T2: Titer der H2SO4 0,5 N (4.4),
m (mg): Probeneinwaage (6.1)
ergibt sich folgende Formel:
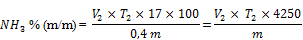
- 8. WIEDERHOLBARKEIT (1)
Bei einem Gehalt von ca. 6% NH3 darf die Differenz zwischen den Ergebnissen zweier parallel durchgeführter Bestimmungen an derselben Probe nicht höher sein als 0,6%.
‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑‑
(1) Siehe ISO-Norm 5725.
Zuletzt aktualisiert am
09.05.2017
Gesetzesnummer
10010899
Dokumentnummer
NOR12138660
alte Dokumentnummer
N8199547967J
Zusatzdokumente: image001, image002
Lizenziert vom RIS (ris.bka.gv.at - CC BY 4.0 DEED)