Anlage 38
NACHWEIS UND BESTIMMUNG VON 2-PHENOXYETHANOL, 1-PHENOXYPROPAN-2-OL, METHYL-, ETHYL-, PROPYL-, BUTYL- UND BENZYL-4-HYDROXYBENZOAT IN KOSMETISCHEN MITTELN
- A. NACHWEIS
- 1. Zweck und Anwendungsbereich
Diese Methode beschreibt ein dünnschichtchromatographisches Verfahren, das in Verbindung mit der in Abschnitt B beschriebenen quantitativen Bestimmung den Nachweis von 2-Phenoxyethanol, 1-Phenoxypropan-2-ol, Methyl-4-hydroxybenzoat, Ethyl-4-hydroxybenzoat, Propyl-4-Hydroxybenzoat, Butyl-4-hydroxybenzoat und Benzyl-4-hydroxybenzoat in kosmetischen Mitteln ermöglicht.
- 2. Kurzbeschreibung
Die Konservierungsstoffe werden mit Aceton aus der angesäuerten Probe des kosmetischen Mittels extrahiert. Nach Filtration wird die Acetonlösung mit Wasser gemischt, und die Fettsäuren werden im alkalischen Medium als ihre Calciumsalze ausgefällt. Die alkalische Aceton-Wasser-Mischung wird mit Diethylether zur Entfernung der lipophilen Stoffe extrahiert. Nach Ansäuern werden die Konservierungsstoffe mit Diethylether extrahiert. Ein aliquoter Teil des Diethyletherextrakts wird auf eine Kieselgel-Dünnschichtplatte aufgetragen. Nach Entwicklung der Dünnschichtplatte wird das erhaltene Chromatogramm unter UV-Licht betrachtet und mit Millon's Reagenz sichtbar gemacht.
- 3. Reagenzien
- 3.1. Allgemeines
Alle Reagenzien müssen analysenrein sein. Wasser muß destilliert sein oder zumindest gleichwertige Reinheit aufweisen.
- 3.2. Aceton
- 3.3. Diethylether
- 3.4. n-Pentan
- 3.5. Methanol
- 3.6. Eisessig
- 3.7. Salzsäure, c(HCl)= 4 mol/l
- 3.8. Kalilauge, c(KOH)= 4 mol/l
- 3.9. Calciumchlorid-Dihydrat (CaCl2
2H2O
- 3.10. Nachweisreagenz: Millon's Reagenz
Millon's Reagenz [vorwiegend Quecksilber(II)-nitrat] ist als gebrauchsfertige Lösung im Handel erhältlich (Fluka 69820).
- 3.11. 2-Phenoxyethanol
- 3.12. 1-Phenoxypropan-2-ol
- 3.13. Methyl-4-hydroxybenzoat (Methylparaben)
- 3.14. Ethyl-4-hyrdoxybenzoat (Ethylparaben)
- 3.15. n-Propyl-4-hydroxybenzoat (Propylparaben)
- 3.16. n-Butyl-4-hydroxybenzoat (Butylparaben)
- 3.17. Benzyl-4-hydroxybenzoat (Benzylparaben)
- 3.18. Vergleichslösungen
Jeweils 0,1%ige (m/V) Lösungen der Vergleichssubstanzen 3.11, 3.12, 3.13, 3.14, 3.15, 3.16 und 3.17 in Methanol
- 3.19. Fließmittel
88 Volumenteile n-Pentan (3.4) werden mit 12 Volumenteilen Eisessig (3.6) gemischt.
- 4. Geräte
Normale Laborausstattung und
- 4.1. Wasserbad, auf 60 °C konstant einstellbar
- 4.2. Entwicklungskammer (nicht mit Filterpapier ausgekleidet)
- 4.3. UV-Lampe, 254 nm
- 4.4. DC-Fertigplatten (200 mm x 200 mm), Sorbensschicht: Kieselgel 60 F254 (Fluoreszensindikator), Schichtdicke: 0,25 mm mit Konzentrierungszone 25 mm x 200 mm (Merck 11798, Darmstadt oder gleichwertiges Erzeugnis)
- 4.5. Trockenschrank, bis auf 105 °C konstant einstellbar
- 4.6. Heißluftfön
- 4.7. Woll-Farbroller, etwa 10 cm breit, Außendurchmesser etwa 3,5 cm. Die Dicke der Wollschicht soll 2 bis 3 mm betragen; gegebenenfalls ist die Wolle zu kürzen. (Siehe Anmerkung unter 5.2).
- 4.8. Erlenmeyerkolben mit Schliffstopfen, 50 ml
- 4.9. Elektrische Heizplatte mit Thermostatregler.
- 5. Durchführung
- 5.1. Vorbereitung der Probe
Ungefähr 1,0 g der Probe wird in einen 50-ml-Erlenmeyerkolben (4.8) mit 4 Tropfen Salzsäure (3.7) und 40 ml Aceton versetzt.
Bei stark basischen Erzeugnissen, wie Feinseife, werden 20 Tropfen Salzsäure hinzugefügt. Der Erlenmeyerkolben wird verschlossen und vorsichtig auf etwa 60 °C erwärmt, damit die Extraktion der Konservierungsstoffe in die Aceton-Phase erleichtert wird, und anschließend eineMinute lang kräftig geschüttelt.
Der pH-Wert der Mischung wird mit pH-Indikatorpapier gemessen und mit Salzsäure auf < 3 eingestellt. Die Mischung wird erneut eine Minute lang kräftig geschüttelt.
Die Mischung wird auf Zimmertemperatur abgekühlt und durch ein Papierfilter in einem Erlenmeyerkolben filtriert. 20 ml des Filtrats werden in einen 200-ml-Erlenmeyerkolben übergeführt mit 60 ml Wasser versetzt und gemischt. Mit Kalilauge (3.8) wird der pH-Wert der Mischung gegen Indikatorpapier auf ungefähr 10 eingestellt.
Nach Zusatz von 1 g Calciumchlorid (3.9) wird die Mischung kräftig geschüttelt und danach durch ein Papierfilter in einen 250-ml-Scheidetrichter, der 75 ml Diethylether enthält, filtriert. Die Mischung wird eine Minute lang kräftig geschüttelt. Nach der Phasentrennung wird die wäßrige Phase in einen 200-ml-Erlenmeyerkolben abgelassen. Mit Hilfe von Indikatorpapier wird der pH-Wert der wäßrigen Lösung mit Salzsäure auf ungefähr 2 eingestellt. Nach Zusatz von 10 ml Diethylether wird die Mischung eine Minute lang kräftig geschüttelt. Nach der Phasentrennung werden etwa 2 ml der Etherphase in ein 5-ml-Probenfläschchen übergeführt.
- 5.2. Dünnschichtchromatographie
Die Dünnschichtplatte (4.4) wird auf die erwärmte Aluminiumplatte (4.9) gelegt. Von jeder Vergleichslösung (3.18) werden 10
l und von der Probelösung (5.1) 100
Erforderlichenfalls kann zur Beschleunigung der Verdunstung des Lösungsmittels ein Luftstrom verwendet werden. Die Dünnschichtplatte wird von der Heizplatte genommen und auf Raumtemperatur abgekühlt. 100 ml des Fließmittels (3.19) werden in die Entwicklungskammer (4.2) gefüllt und die Dünnschichtplatte sofort in die ungesättigte Kammer gestellt und bei Raumtemperatur bis zu einer Höhe von etwa 15 cm entwickelt. Nach Entnahme aus der Kammer wird die Platte im Heißluftstrom (Fön) getrocknet.
Die Dünnschichtplatte wird im UV-Licht (4.3) betrachtet, und die Positionen der Flecke werden markiert. Anschließend wird die Platte im Trockenschrank (4.5) 30 Minuten lang bei 100 °C erwärmt, um überschüssige Essigsäure zu entfernen. Die Konservierungsstoffe werden auf der Dünnschichtplatte durch Eintauchen des Farbrollers (4.7) in das Millon's Reagenz (3.10) und Überstreichen derPlatte bis zur gleichmäßigen Benetzung sichtbar gemacht.
Hinweis: Alternativ können die Flecke durch sorgfältiges Auftragen eines Tropfens Millon's Reagenz auf die im UV-Licht markierten Flecke sichtbar gemacht werden.
Die Ester der 4-Hydroxybenzoesäure erscheinen als rote, 2-Phenoxyethanol und 1-Phenoxy-propan-2-ol als gelbe Flecke. Zu beachten ist allerdings, daß die 4-Hydroxybenzoesäure selbst, die in den Proben als Konservierungsstoff oder als Zersetzungsprodukt ihrer Ester anwesend sein kann, ebenfalls als roter Fleck erscheint. (Siehe unter 7.3 und 7.4).
- 6. Auswertung
Der Rf-Wert wird für jeden Fleck berechnet. Die für die Probelösung erhaltenen Flecke werden mit denen der Vergleichslösungen im Hinblick auf ihre Rf-Werte, ihr Verhalten im UV-Licht und ihre Färbung nach Sichtbarmachung mit dem Reagenz verglichen. Im Ergebnis dieses Vergleichs wird eine vorläufige Schlußfolgerung gezogen, welche Konservierungsstoffe vorliegen. Zur Bestätigung sollte das im Abschnitt B beschriebene HPLC-Verfahren durchgeführt werden. Der Nachweis der Konservierungsstoffe wird abschließend durch Kombination der Ergebnisse der DC und der HPLC geführt.
- 7. Bemerkungen
- 7.1. Millon's Reagenz wird wegen seiner Toxizität am besten nach einem der beschriebenen Verfahren angewandt. Sprühen ist nicht zu empfehlen.
- 7.2. Andere Hydroxylgruppen enthaltende Verbindungen können ebenfalls mit Millon's Reagenz angefärbt werden. Eine Übersicht über Färbungen und Rf-Werte für eine Reihe von Konservierungsstoffen findet man in: N. De Kruijf, M.A.H. Rijk, L.A. Pranoto-Soetardhi und A. Schouten: Determination of preservatives in cosmetic products I: Thin layer chromatographic procedure for the identification of preservatives in cosmetic products (J. Chromatogr., 410, 395-411 (1987)).
- 7.3. Die in folgender Tabelle aufgelisteten Rf-Werte geben Hinweise auf die zu erwartenden Werte.
Substanz | hRf | Anfärbung |
4-Hydroxybenzoesäure | 11 | rot |
Methylparaben | 12 | rot |
Ethylparaben | 17 | rot |
Propylparaben | 21 | rot |
Butylparaben | 26 | rot |
Benzylparaben | 16 | rot |
2-Phenoxyethanol | 29 | gelb |
1-Phenoxypropan-2-ol | 50 | gelb |
- 7.4. Keine Trennung erreicht man für 4-Hydroxybenzoesäure und Methylparaben bzw. für Benzylparaben und Ethylparaben. Der Nachweis dieser Substanzen sollte mittels HPLC nach Abschnitt B durch Vergleich der Retentionszeiten der Probenpeaks mit denen der Standards bestätigt werden.
- 1. Zweck und Anwendungsbereich
Diese Methode beschreibt ein Verfahren zur quantitativen Bestimmung von 2-Phenoxyethanol, 1-Phenoxypropan-2-ol, Methyl-4-hydroxybenzoat, Ethyl-4-hydroxybenzoat, Propyl-4-hydroxybenzoat, Butyl-4-hydroxybenzoat und Benzyl-4-hydroxybenzoat in kosmetischen Mitteln.
- 2. Definition
Die nach diesem Verfahren ermittelten Gehalte an Konservierungsstoffen werden in Prozent (% m/m) angegeben.
- 3. Kurzbeschreibung
Nach dem Ansäuern mit Schwefelsäure wird die Probe in einer Mischung aus Ethanol und Wasser suspendiert. Die Mischung wird vorsichtig bis zum Schmelzen der Lipidphase erwärmt, um die quantitative Extraktion zu gewährleisten.
Nach der Filtration wird der Gehalt an Konservierungsstoffen durch Hochleistungsflüssigkeitschromatographie (HPLC) mit Umkehrphase und Isopropyl-4-hydroxybenzoat als innerem Standard bestimmt.
- 4. Reagenzien
- 4.1. Allgemeines
Alle Reagenzien müssen analysenrein und, wo erforderlich, für die HPLC geeignet sein. Wasser muß destilliert sein oder zumindest gleichwertige Reinheit aufweisen.
- 4.2. Absolutes Ethanol
- 4.3. 2-Phenoxyethanol
- 4.4. 1-Phenoxypropan-2-ol
- 4.5. Methyl-4-hydroxybenzoat (Methylparaben)
- 4.6. Ethyl-4-hydroxybenzoat (Ethylparaben)
- 4.7. n-Propyl-4-hydroxybenzoat (Propylparaben)
- 4.8. Isopropyl-4-hydroxybenzoat (Isopropylparaben)
- 4.9. n-Butyl-4-hydroxybenzoat (Butylparaben)
- 4.10. Benzyl-4-hydroxybenzoat (Benzylparaben)
- 4.11. Tetrahydrofuran
- 4.12. Methanol
- 4.13. Acetonitril
- 4.14. Schwefelsäure, c(H2SO4)= 2 mol/l
- 4.15. Ethanol/Wasser-Mischung
9 Volumenteile Ethanol (4.2) werden mit 1 Volumenteil Wasser gemischt.
- 4.16. Lösung des inneren Standards
Ungefähr 0,25 g Isopropylparaben (4.8) werden in einen 500-ml-Meßkolben eingewogen, in der Ethanol/Wasser-Mischung (4.15) gelöst und zur Marke aufgefüllt.
- 4.17. Mobile Phase
5 Volumenteile Tetrahydrofuran, 60 Volumenteile Wasser, 10 Volumenteile Methanol und 25 Volumenteile Acetonitril werden gemischt.
- 4.18. Stammlösung der Konservierungsstoffe
Ungefähr 0,2 g 2-Phenoxyethanol, 0,2 g 1-Phenoxypropan-2-ol, 0,05 g Methylparaben, 0,05 g Ethylparaben, 0,05 g Propylparaben, 0,05 g Butylparaben und 0,025 g Benzylparaben werden in einen 100-ml-Meßkolben genau eingewogen, gelöst und mit der Ethanol/Wasser-Mischung zur Marke aufgefüllt.
Die Lösung wird im Kühlschrank aufbewahrt und ist eine Woche lang haltbar.
- 4.19. Standardlösungen der Konservierungsstoffe
Von der Stammlösung (4.18) werden 20,00 ml, 10,00 ml, 5,00 ml, 2,00 ml bzw. 1,00 ml in eine Reihe von 50-ml-Meßkolben pipettiert. Nach Zusatz von jeweils 10,00 ml der Lösung des inneren Standards (4.16) und 1,0 ml Schwefelsäure (4.14) wird mit der Ethanol/Wasser-Mischung zur Marke aufgefüllt und gemischt. Die Lösungen sind frisch herzustellen.
- 5. Geräte
Normale Laborausstattung und
- 5.1. Wasserbad, auf 60 °C
1 °C einstellbar
- 5.2. Hochleistungsflüssigkeitschromatograph mit UV-Dektektor (Anm.: richtig: Detektor), Wellenlänge 280 nm
- 5.3. Analytische Trennsäule
Material: Edelstahl, Länge: 12,5 cm oder 25 cm, Innendurchmesser: 4,6 mm, aktiver Festkörper: Mit Octadecylgruppen modifiziertes Kieselgel (Nucleosil 5 Cl8 oder gleichwertiges Erzeugnis (siehe 10.1))
- 5.4. Erlenmeyerkolben mit Schliffstopfen, 100 ml
- 5.5. Siedesteinchen (Karborund), Größe 2 bis 4 mm, oder gleichwertiges Material
- 6. Durchführung
- 6.1. Vorbereitung der Probe
- 6.1.1. Vorbereitung der Probe ohne Zugabe des inneren Standards
Ungefähr 1 g der Probe wird in einen 100-ml-Erlenmeyerkolben eingewogen. Nach Zusatz von 1,00 ml Schwefelsäure (4.14) und 50,0 ml der Ethanol/Wasser-Mischung (4.15) sowie ungefähr 1 g Siedesteinchen (5.5) wird der verschlossene Kolben kräftig geschüttelt, bis eine homogene Suspension entstanden ist, mindestens jedoch 1 Minute. Zur Verbesserung der Extraktion wird der Kolben 5 Minuten im Wasserbad (5.1) auf 60 °C
1 °C erwärmt.
Danach wird der Kolben sofort unter fließendem Wasser abgekühlt und anschließend eine Stunde lang im Kühlschrank aufbewahrt. Der Extrakt wird durch einen Papierfilter filtriert. Etwa 2 ml des Filtrats werden in ein 5-ml-Probenfläschchen übergeführt und im Kühlschrank aufbewahrt. Die HPLC-Bestimmung ist innerhalb von 24 Stunden nach Herstellung der Probelösung durchzuführen.
- 6.1.2. Vorbereitung der Probe unter Zugabe des inneren Standards
1,0 g
Nach Zusatz von 1,00 ml Schwefelsäure und 40,0 ml der Ethanol/Wasser-Mischung sowie ungefähr 1 g Siedesteinchen und genau 10,00 ml der Lösung des inneren Standards (4.16) wird der verschlossene Kolben kräftig geschüttelt, bis eine homogene Suspension entstanden ist, mindestens jedoch 1 Minute. Zur Verbesserung der Extraktion wird der Kolben 5 Minuten im Wasserbad auf 60 °C
Danach wird der Kolben sofort unter fließendem kaltem Wasser abgekühlt und anschließend eine Stunde lang im Kühlschrank aufbewahrt. Der Extrakt wird durch ein Papierfilter filtriert.
Etwa 2 ml des Filtrats werden in ein 5-ml-Probenfläschchen übergeführt (Prüflösung) und im Kühlschrank aufbewahrt. Die HPLC-Bestimmung ist innerhalb von 24 Stunden nach Herstellung der Prüflösung durchzuführen.
- 6.2. Hochleistungsflüssigkeitschromatographie
- 6.2.1. Bedingungen für die Chromatographie
- Mobile Phase: Tetrahydrofuran/Wasser/Methanol/Acetonitril-Mischung (4.17)
- Volumenstrom der mobilen Phase: 1,5 ml/min
- Detektionswellenlänge: 280 nm
- 6.2.2. Kalibrierung
10
- 6.2.3. Bestimmung
Jeweils 10
Falls das Chromatogramm der Probelösung (6.1.1) keinen Peak mit der Retentionszeit des empfohlenen inneren Standards Isopropylparaben zeigt, werden 10
Falls im Chromatogramm der Probelösung (6.1.1) ein störender Peak erscheint, der etwa dieselbe Retentionszeit wie Isopropylparaben aufweist, ist ein anderer geeigneter innerer Standard zu wählen.
Als innerer Standard kann einer der zu bestimmenden Konservierungsstoffe verwendet werden, der nicht auf dem Chromatogramm der Probelösung erscheint.
Es wird das Verhältnis zwischen den Peakhöhen der zu bestimmenden Konservierungsstoffe und der des inneren Standards ermittelt.
Für die Standardlösung soll die Eichkurve eine Gerade bilden.
Die Chromatogramme der Standardlösungen und der Probelösung sollen folgende Bedingungen erfüllen:
- Peakauflösung p: Die Peakauflösung des am schlechtesten getrennten Paares soll mindestens 0,90 betragen (zur Definition der Peakauflösung s. Abb. 1).
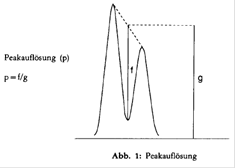
Falls die erforderliche Auflösung nicht erreicht wird, verwendet man entweder eine leistungsfähigere Säule oder verändert die Zusammensetzung der mobilen Phase, bis die Bedingung erfüllt ist.
- Peakasymmetrie: Der Asymmetriefaktor As aller interessierenden Peaks soll zwischen 0,9 und 1,5 liegen (zur Definition des Asymmetriefaktors s. Abb. 2). Bei der Aufzeichnung des Chromatogramms zur Bestimmung der Peakasymmetrie sollte der Papiervorschuß mindestens 2 cm/min betragen.
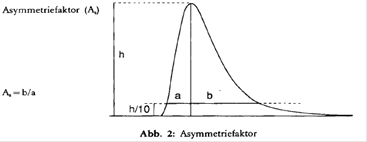
- Basislinie: Die Basislinie sollte stabil sein.
- 7. Berechnung
Unter Verwendung der Verhältnisse zwischen den Peakhöhen der zu bestimmenden Konservierungsstoffe und der Peakhöhe des inneren Standards wird die Konzentration der Konservierungsstoffe in der Prüflösung aus der Kalibierkurve (6.2.2) abgelesen bzw. aus der Gleichung für die Regressionsgerade berechnet. Der Gehalt wi an 2-Phenoxyethanol, 1-Phenoxypropan-2-ol, Methyl-4-hydroxybenzoat, Ethyl-4-hydroxybenzoat, Propyl-4-hydroxybenzoat, Butyl-4-hydroxybenzoat und Benzyl-4-hydroxybenzoat in Prozent (% m/m) der Probe wird nach folgender Formel berechnet:
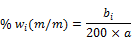
wobei:
- bi = aus der Kalibrierkurve abgelesene Konzentration des Konservierungsstoffes i in der Prüflösung (6.1.2) in Mikrogramm je Milliliter bzw. der aus der Gleichung für die Regressionsgerade berechnete Wert
- a = Einwaage der untersuchten Probe (6.1.2) in Gramm
- 8. Wiederholbarkeit (1)
Siehe Bemerkungen unter 10.5
- 9. Vergleichbarkeit (1)
Siehe Bemerkungen unter 10.5
- 10. Bemerkungen
- 10.1. Stationäre Phase
Des Retentionsverhalten der Substanzen ist stark abhängig vom Typ, der Handelsmarke und der Vorgeschichte der Trennsäule (stationäre Phase). Ob eine Säule für die Trennung der zu bestimmenden Konservierungsstoffe geeignet ist, kann aus den für die Standardlösungen gewonnenen Ergebnissen geschlossen werden (siehe Bemerkungen unter 6.2.3). Neben der vorgeschlagenen Handelsmarke sind auch die Füllmaterialien Hypersil ODS und Zorbax ODS als stationäre Phasen geeignet.
Alternativ kann auch die Zusammensetzung der mobilen Phase zur Erzielung der geforderten Auflösung optimiert werden.
- 10.2. Detektionswellenlänge
Der Robustheitstest (ruggedness test) mit der beschriebenen Methode hat ergeben, daß eine geringfügige Änderung der Detektionswellenlänge sich signifikant auf die Ergebnisse der Bestimmung auswirken kann. Bei der Analyse muß daher dieser Parameter sorgfältig kontrolliert werden.
- 10.3. Interferenzen
Unter den Bedingungen der beschriebenen Methode werden auch viele andere Substanzen, wie weitere Konservierungsstoffe und kosmetische Zusatzstoffe, eluiert. Retentionszeiten für eine Vielzahl der im Anhang VI der Richtlinie des Rates über kosmetische Mittel erwähnten Konservierungsstoffe findet man in N. De Kruijf, M.A.H. Rijk, L.A. Pranato-Soetardhi und A. Schouten:
Determination of preservatives in cosmetic products II. High performance liquid chromatographic identification (J. Chromatogr. 469, 317-328 (1989)).
- 10.4. Zum Schutz der Trennsäule empfiehlt sich die Verwendung einer geeigneten Vorsäule.
- 10.5. Die Methode ist in einem Ringversuch unter Beteiligung von 9 Laboratorien an drei kosmetischen Erzeugnissen untersucht worden. In der folgenden Tabelle sind für jede der drei Proben der Mittelwert m in Prozent (% m/m), die Wiederholbarkeit r und die Vergleichbarkeit R für die jeweils ermittelten Analyten aufgelistet:
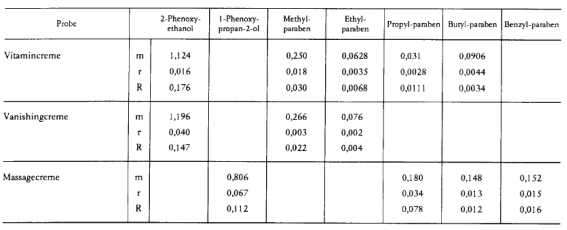
___________
(1) ISO 5725.
Zuletzt aktualisiert am
09.05.2017
Gesetzesnummer
10010899
Dokumentnummer
NOR12140907
alte Dokumentnummer
N8199712667Y
Zusatzdokumente: image001, image002, image003, image004, image005, image006, image007, image008
Lizenziert vom RIS (ris.bka.gv.at - CC BY 4.0 DEED)