Anlage 36
NACHWEIS UND BESTIMMUNG VON BENZOESÄURE, 4‑HYDROXYBENZOESÄURE, SORBINSÄURE, SALICYLSÄURE UND PROPIONSÄURE IN KOSMETISCHEN MITTELN
- 1. Zweck und Anwendungsbereich
Diese Methode beschreibt ein Verfahren zum Nachweis und zur Bestimmung von Benzoesäure, 4-Hydroxybenzoesäure, Sorbinsäure, Salicylsäure und Propionsäure in kosmetischen Mitteln. Die Methode gliedert sich in Verfahren
- zum Nachweis dieser Konservierungsstoffe,
- zur Bestimmung von Benzoesäure, 4-Hydroxybenzoesäure, Sorbinsäure und Salicylsäure,
- zur Bestimmung von Propionsäure.
- 2. Definition
Der nach diesen Verfahren ermittelte Gehalt an Benzoesäure, 4-Hydroxybenzoesäure, Sorbinsäure, Salicylsäure und Propionsäure wird in Prozent (% m/m) an freier Säure angegeben.
- A. NACHWEIS
- 1. Kurzbeschreibung
Im Anschluß an eine Säure/Base-Extraktion der Konservierungsstoffe wird der Extrakt mittels DC und Reaktionschromatographie („On-plate-Derivatisierung“) analysiert. In Abhängigkeit vom Ergebnis erfolgt eine Bestätigung des Nachweises durch HPLC oder, im Fall von Propionsäure, durch GC.
- 2. Reagenzien
- 2.1. Alle Reagenzien müssen analysenrein sein. Wasser muß destilliert sein oder zumindest gleichwertige Reinheit aufweisen.
- 2.2. Aceton
- 2.3. Diethylether
- 2.4. Acetonitril
- 2.5. Toluen
- 2.6. n-Hexan
- 2.7. Flüssiges Paraffin
- 2.8. Salzsäure, 4 M
- 2.9. Kalilauge, 4 M
- 2.10. Calciumchlorid, CaCl2
2 H2O
- 2.11. Lithiumcarbonat, Li2CO3
- 2.12. 2-Brom-2'-acetonaphthon
- 2.13. 4-Hydroxybenzoesäure
- 2.14. Salicylsäure
- 2.15. Benzoesäure
- 2.16. Sorbinsäure
- 2.17. Propionsäure
- 2.18. Vergleichslösungen
Jeweils 0,1%ige (m/V) Lösungen (100 mg/100 ml) der fünf Konservierungsstoffe (2.13 bis 2.17) im Diethylether.
- 2.19. Derivatisierungsreagenz
0,5%ige (m/V) Lösung von 2-Brom-2'-acetonaphthon (2.12) in Acetonitril (2.4). Die Lösung muß wöchentlich frisch hergestellt und im Kühlschrank aufbewahrt werden.
- 2.20. Katalysator-Lösung
0,3%ige (m/V) Lösung von Lithiumcarbonat (2.11) in Wasser (300 mg/100 ml). Diese Lösung muß frisch hergestellt werden.
- 2.21. Fließmittel
Toluen (2.5)/Aceton (2.2), 20 + 0,5 (V + V)
- 2.22. Tauch-Lösung
Flüssiges Paraffin (2.7)/n-Hexan (2.6), 1 + 2 (V + V)
- 3. Geräte
Normale Laborausstattung und
- 3.1. Wasserbad, auf 60 °C konstant einstellbar
- 3.2. Chromatographiekammer
- 3.3. UV-Lampe, 254 und 366 nm
- 3.4. DC-Fertigplatten (200 mm x 200 mm)
Sorbensschicht: Kieselgel 60 ohne Fluoreszenzindikator
Schichtdicke: 0,25 mm mit Konzentrierungszone 25 mm x 200 mm (Merck 11845 oder gleichwertiges Erzeugnis)
- 3.5. Mikroliterspritze, 10
l
- 3.6. Mikroliterspritze, 25
- 3.7. Trockenschrank, bis auf 105 °C konstant einstellbar
- 3.8. Erlenmeyerkolben mit Schliffstopfen, 50 ml und 200 ml
- 3.9. Papierfilter, Durchmesser 90 mm (Schleicher und Schüll, Weißband Nr. 5892, oder gleichwertiges Erzeugnis)
- 3.10. Universal pH-Indikatorpapier, pH 1-11
- 3.11. Probenfläschchen, 5 ml
- 3.12. Rotationsverdampfer
- 3.13. Heizplatte
- 4. Durchführung
- 4.1. Vorbereitung der Probe
Ungefähr 1,0 g der Probe wird in einen 50-ml-Erlenmeyerkolben (3.8) mit 4 Tropfen 4 M Salzsäure (2.8) und 40 ml Aceton (2.2) versetzt.
Bei stark basischen Erzeugnissen, wie Feinseife, werden 20 Tropfen 4 M Salzsäure (2.8) hinzugefügt. Der mit Indikatorpapier (3.10) gemessene pH-Wert soll ungefähr 2 betragen.
Der Erlenmeyerkolben wird verschlossen und eine Minute lang kräftig geschüttelt. Notfalls kann die Mischung vorsichtig bis auf 60 °C erwärmt werden, damit die Fettphase schmilzt und die Extraktion der Konservierungsstoffe in die Aceton-Phase erleichtert wird.
Die Mischung wird auf Zimmertemperatur abgekühlt und durch ein Papierfilter (3.9) filtriert. 20 ml des Filtrats werden in einen 200-ml-Erlenmeyerkolben übergeführt, mit 20 ml Wasser versetzt und gemischt. Mit 4 M Kalilauge (2.9) wird der pH-Wert der Mischung gegen Idikatorpapier (3.10) auf ungefähr 10 eingestellt.
Nach Zusatz von 1 g Calciumchlorid (2.10) wird die Mischung kräftig geschüttelt und danach durch ein Papierfilter (3.9) in einen 250-ml-Scheidetrichter, der 75 ml Diethylether (2.3) enthält, filtriert. Die Mischung wird eine Minute lang kräftig geschüttelt. Nach der Phasentrennung wird die wäßrige Phase in einen weiteren 250-ml-Scheidetrichter abgelassen und die Etherphase verworfen. Mit Hilfe von Indikatorpapier (3.10) wird der pH-Wert der wäßrigen Lösung mit 4 M Salzsäure (2.8) auf ungefähr 2 eingestellt. Nach Zusatz von 10 ml Diethylether (2.3) wird die Mischung eine Minute lang kräftig geschüttelt. Nach der Phasentrennung wird die Etherphase in einen Rotationsverdampfer (3.12) übergeführt und die wäßrige Phase verworfen.
Die Etherphase wird fast zur Trockne eingedampft und der Rückstand in 1 ml Diethylether (2.3) aufgenommen. Diese Lösung wird in ein Probenfläschchen (3.11) übergeführt.
- 4.2. Dünnschichtchromatographie
Entsprechend der Anzahl der zu chromatographierenden Vergleichs- und Probelösungen werden auf die Startlinie in der Konzentrierungszone der Dünnschichtplatte in gleichen Abständen je 3
Die Dünnschichtplatte wird im Trockenschrank (3.7) 45 Minuten lang bei 80 °C erwärmt. Nach dem Abkühlen wird die Dünnschichtplatte in die vorher für 15 Minuten mit dem Fließmittel (2.21) äquilibrierte Chromatographiekammer (3.2) (ohne Filterpapier) gestellt und bis zu einer Höhe von 15 cm entwickelt (Zeitdauer etwa 80 Minuten).
Die Dünnschichtplatte wird im Kaltluftstrom getrocknet und im UV-Licht (3.3) betrachtet. Die Fluoreszenz der schwachen Flecke kann durch Tauchen der Dünnschichtplatte in flüssiges Paraffin/n-Hexan (2.22) verstärkt werden.
- 5. Auswertung
Der Rf-Wert wird für jeden Fleck berechnet. Die für die Probelösung erhaltenen Flecke werden mit denen der Vergleichslösungen im Hinblick auf ihre Rf-Werte und ihr Verhalten im UV-Licht verglichen.
Im Ergebnis dieses Vergleichs wird eine vorläufige Schlußfolgerung gezogen, ob und welche Konservierungsstoffe vorliegen.
Man führt die im folgenden Abschnitt B beschriebene HPLC oder, im Fall der Propionsäure, die im Abschnitt C beschriebene GC durch und vergleicht die Retentionszeiten der Probenpeaks mit denen der Vergleichslösungen. Der Nachweis der Konservierungsstoffe wird abschließend durch Kombination der Ergebnisse der DC und der HPLC bzw. GC geführt.
- B. BESTIMMUNG VON BENZOESÄURE, 4-HYDROXYBENZOESÄURE, SORBINSÄURE UND SALICYLSÄURE
- 1. Kurzbeschreibung
Nach dem Ansäuern wird die Probe mit einer Mischung aus Ethanol und Wasser extrahiert. Nach der Filtration wird der Gehalt an Konservierungsstoffen durch Hochleistungsflüssigkeitschromatographie (HPLC) bestimmt.
- 2. Reagenzien
- 2.1. Alle Reagenzien müssen analysenrein und, wo erforderlich, für die HPLC geeignet sein. Wasser muß destilliert sein oder zumindest gleichwertige Reinheit aufweisen.
- 2.2. Absolutes Ethanol
- 2.3. 4-Hydroxybenzoesäure
- 2.4. Salicylsäure
- 2.5. Benzoesäure
- 2.6. Sorbinsäure
- 2.7. Natriumacetat, CH3COONa
- 2.8. Essigsäure,
d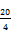
- 2.9. Acetonitril
- 2.10. Schwefelsäure, 2 M
- 2.11. Kalilauge, 0,2 M
- 2.12. 2-Methoxybenzoesäure
- 2.13. Ethanol/Wasser-Mischung
9 Volumenteile Ethanol (2.2) werden mit 1 Volumenteil Wasser (2.1) gemischt.
- 2.14. Lösung des inneren Standards
Ungefähr 1 g 2-Methoxybenzoesäure (2.12) wird in 500 ml Ethanol/Wasser-Mischung (2.13) gelöst.
- 2.15. Mobile Phase
- 2.15.1. Acetatpuffer: 6,35 g Natriumacetat (2.7) und 20,0 ml Essigsäure (2.8) werden in 1 l Wasser gelöst.
- 2.15.2. Mobile Phase: 9 Volumenteile Acetatpuffer (2.15.1) und 1 Volumenteil Acetonitril (2.9) werden gemischt.
- 2.16. Stammlösung der Konservierungsstoffe
Ungefähr 0,05 g 4-Hydroxybenzoesäure (2.3), 0,2 g Salicylsäure (2.4), 0,2 g Benzoesäure (2.5) und 0,05 g Sorbinsäure (2.6) werden in einen 50-ml-Meßkolben genau eingewogen und mit der Ethanol/Wasser-Mischung (2.13) zur Marke aufgefüllt. Die Lösung wird im Kühlschrank aufbewahrt und ist eine Woche lang haltbar.
- 2.17. Standardlösungen der Konservierungsstoffe
Von der Stammlösung (2.16) werden 8,00, 4,00, 2,00, 1,00 bzw. 0,50 ml in eine Reihe von 20‑ml-Meßkolben pipettiert. Nach Zusatz von jeweils 10,00 ml der Lösung des inneren Standards (2.14) mittels Pipette und 0,5 ml 2 M Schwefelsäure (2.10) wird mit der Ethanol/Wasser-Mischung (2.13) zur Marke aufgefüllt und gemischt. Die Lösungen sind frisch herzustellen.
- 3. Geräte
Normale Laborausstattung und
- 3.1. Wasserbad, auf 60 °C konstant einstellbar
- 3.2. Hochleistungsflüssigkeitschromatograph mit UV-Detektor mit variabler Wellenlänge und mit 10
- 3.3. Analytische Trennsäule
Material: Edelstahl
Länge: 12,5 bis 25 cm
Innendurchmesser: 4,6 mm
Aktiver Festkörper: Mit Octadecylgruppen modifiziertes Kieselgel (Nucleosil 5 C18 oder gleichwertiges Erzeugnis)
- 3.4. Papierfilter, Durchmesser 90 mm (Schleicher und Schüll, Weißband Nr. 5892, oder gleichwertiges Erzeugnis)
- 3.5. Erlenmeyerkolben, 50 ml
- 3.6. Probenfläschchen, 5 ml
- 3.7. Siedesteinchen (Karborund), Größe 2 bis 4 mm, oder gleichwertiges Material
- 4. Durchführung
- 4.1. Vorbereitung der Probe
- 4.1.1. Vorbereitung der Probe ohne Zugabe des inneren Standards 1 g der Probe wird in einen 50-ml-Erlenmeyerkolben (3.5) eingewogen. Nach Zusatz von 1,00 ml 2 M Schwefelsäure (2.10) und 40,0 ml der Ethanol/Wasser-Mischung (2.13) sowie ungefähr 1 g Siedesteinchen (3.7) wird der verschlossene Kolben kräftig geschüttelt, bis eine homogene Suspension entstanden ist, mindestens jedoch 1 Minute. Zur Verbesserung der Extraktion wird der Kolben genau 5 Minuten im Wasserbad (3.1) auf 60 °C erwärmt. Danach wird der Kolben sofort unter fließendem kaltem Wasser abgekühlt und anschließend eine Stunde lang bei 5 °C aufbewahrt. Der Extrakt wird durch ein Papierfilter (3.4) filtriert. Etwa 2 ml des Filtrats werden in ein Probenfläschchen (3.6) übergeführt und bei 5 °C aufbewahrt.
Die HPLC-Bestimmung ist innerhalb von 24 Stunden nach Herstellung der Probelösung durchzuführen.
- 4.1.2. Vorbereitung der Probe unter Zugabe des inneren Standards
1 g
0,1 g (a Gramm) der Probe wird auf drei Dezimalstellen genau in einen 50-ml-Erlenmeyerkolben (3.5) eingewogen. Nach Zusatz von 1,00 ml 2 M Schwefelsäure (2.10) und 30,0 ml der Ethanol/Wasser-Mischung (2.13) sowie ungefähr 1 g Siedesteinchen (3.7) und 10,00 ml der Lösung des inneren Standards (2.14) wird der verschlossene Kolben kräftig geschüttelt, bis eine homogene Suspension entstanden ist, mindestens jedoch 1 Minute. Zur Verbesserung der Extraktion wird der Kolben genau 5 Minuten im Wasserbad (3.1) auf 60 °C erwärmt. Danach wird der Kolben sofort unter fließendem kaltem Wasser abgekühlt und anschließend eine Stunde lang bei 5 °C aufbewahrt. Der Extrakt wird durch ein Papierfilter (3.4) filtriert. Etwa 2 ml des Filtrats werden in ein Probenfläschchen (3.6) übergeführt und bei 5 °C aufbewahrt.
Die HPLC-Bestimmung ist innerhalb von 24 Stunden nach Herstellung der Probelösung durchzuführen.
- 4.2. Hochleistungsflüssigkeitschromatographie
Bedingungen für die Chromatographie
Mobile Phase: Acetonitril/Acetatpuffer (2.15.2)
Volumenstrom der mobilen Phase: 2,0 ml/min
Detektionswellenlänge: 240 nm
- 4.2.1. Kalibrierung
10
Für jede Standardlösung wird das Verhältnis zwischen der Peakhöhe des zu bestimmenden Konservierungsstoffes und der des inneren Standards ermittelt. Es wird eine Kalibrierkurve gezeichnet, indem das Peakhöhenverhältnis in Abhängigkeit von der Konzentration des Konservierungsstoffes aufgetragen wird, oder es wird die Regressionsgerade berechnet. Man vergewissert sich, daß sich für die Standardlösungen eine lineare Kurve ergibt.
- 4.2.2. Bestimmung
Jeweils 10
Falls im Chromatogramm der Probelösung (4.1.1) ein störender Peak erscheint, der dieselbe Retentionszeit wie 2-Methoxybenzoesäure aufweist, ist ein anderer geeigneter innerer Standard zu wählen. Als innerer Standard kann einer der zu bestimmenden Konservierungsstoffe verwendet werden, der nicht auf dem Chromatogramm der Probelösung erscheint. Die Chromatogramme der Standardlösung und der Probelösung sollen folgende Bedingungen erfüllen:
- Peakauflösung: Die Peakauflösung soll mindestens 0,90 betragen (zur Definition der Peakauflösung siehe Abbildung 1).
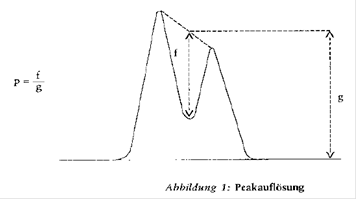
Falls die erforderliche Auflösung nicht erreicht wird, verwendet man entweder eine leistungsfähigere Säule oder verändert die Zusammensetzung der mobilen Phase, bis die Bedingung erfüllt ist.
- Peakasymmetrie: Der Asymmetriefaktor AS soll zwischen 0,9 und 1,5 liegen (zur Definition des Asymmetriefaktors siehe Abbildung 2). Bei der Aufzeichnung des Chromatogramms zur Bestimmung der Peakasymmetrie sollte der Papiervorschub mindestens 2 cm/min betragen.
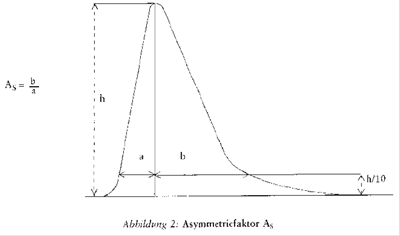
- Basislinie: Die Basislinie sollte stabil sein.
- 5. Berechnung
Unter Verwendung der Verhältnisse zwischen den Peakhöhen der zu bestimmenden Konservierungsstoffe und der Peakhöhe der 2-Methoxybenzoesäure (innerer Standard) wird die Konzentration der Konservierungsstoffe aus der Kalibrierkurve abgelesen bzw. aus der Gleichung für die Regressionsgerade berechnet. Der Gehalt an Benzoesäure, 4‑Hydroxybenzoesäure, Sorbinsäure und Salicylsäure in Prozent (xi) der Probe wird nach folgender Formel berechnet:
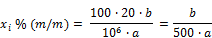
a = | Einwaage der untersuchten Probe (4.1.2) in Gramm |
b = | aus der Kalibrierkurve abgelesene Konzentration des Konservierungsstoffes in der Probelösung (4.1.2) in Mikrogramm je Milliliter bzw. der aus der Gleichung für die Regressionsgerade berechnete Wert |
- 6. Wiederholbarkeit (1)
Bei einem 4-Hydroxybenzoesäuregehalt von 0,40% (m/m) dürfen die Ergebnisse zweier an derselben Probe parallel durchgeführter Bestimmungen um nicht mehr als 0,035% (m/m) voneinander abweichen.
Bei einem Benzoesäuregehalt von 0,50% (m/m) dürfen die Ergebnisse zweier an derselben Probe parallel durchgeführter Bestimmungen um nicht mehr als 0,05% (m/m) voneinander abweichen.
Bei einem Salicylsäuregehalt von 0,50% (m/m) dürfen die Ergebnisse zweier an derselben Probe parallel durchgeführter Bestimmungen um nicht mehr als 0,045% (m/m) voneinander abweichen.
Bei einem Sorbinsäuregehalt von 0,60% (m/m) dürfen die Ergebnisse zweier an derselben Probe parallel durchgeführter Bestimmungen um nicht mehr als 0,035% (m/m) voneinander abweichen.
- 7. Bemerkungen
- 7.1. Der Robustheitstest (ruggedness test) mit dieser Methode ergab, daß die für die Extraktion der Konservierungsstoffe vorgeschriebene Menge an Schwefelsäure das Ergebnis beeinflussen kann und die Einwaage der Probe in den angegebenen Grenzen erfolgen muß.
- 7.2. Falls erforderlich, kann eine geeignete Vorsäule verwendet werden.
- C. BESTIMMUNG VON PROPIONSÄURE
- 1. Zweck und Anwendungsbereich
Diese Methode bechreibt (Anm.: richtig: beschreibt) ein Verfahren zur Bestimmung von Propionsäure bis zu einer Höchstkonzentration von 2% (m/m) in kosmetischen Mitteln.
- 2. Definition
Der nach diesem Verfahren ermittelte Gehalt an Propionsäure wird in Prozent (% m/m) des Erzeugnisses angegeben.
- 3. Kurzbeschreibung
Nach Extraktion der Propionsäure aus dem Erzeugnis wird die Bestimmung mittels Gaschromatographie unter Verwendung von 2-Methylpropionsäure als innerer Standard durchgeführt.
- 4. Reagenzien
Alle Reagenzien müssen analysenrein sein. Wasser muß destilliert sein oder zumindest gleichwertige Reinheit aufweisen.
- 4.1. Ethanol, 96% (V/V)
- 4.2. Propionsäure
- 4.3. 2-Methylpropionsäure
- 4.4. Orthophosphorsäure, 10% (m/V)
- 4.5. Propionsäure-Standardlösung
Ungefähr 1,00 g (p Gramm) Propionsäure wird genau in einen 50-ml-Meßkolben eingewogen und mit Ethanol (4.1) zur Marke aufgefüllt.
- 4.6. Lösung des inneren Standards
Ungefähr 1,00 g (e Gramm) 2-Methylpropionsäure wird genau in einen 50-ml-Meßkolben eingewogen und mit Ethanol (4.1) zur Marke aufgefüllt.
- 5. Geräte
- 5.1. Normale Laborausstattung und
- 5.2. Gaschromatograph mit Flammenionisationsdetektor
- 5.3. Reagenzglas, 20 ml, mit Schliffstopfen
- 5.4. Wasserbad, auf 60 °C konstant einstellbar
- 5.5. 10-ml-Glasspritze mit Membranfiltereinheit, 0,45
- 6. Durchführung
- 6.1. Vorbereitung der Probe
- 6.1.1. Vorbereitung der Probe ohne Zugabe des inneren Standards
1 g der Probe wird in ein Reagenzglas mit Schliffstopfen (5.3) eingewogen. Nach Zusatz von 0,50 ml Phosphorsäure (4.4) und 9,5 ml Ethanol (4.1) wird das verschlossene Reagenzglas kräftig geschüttelt. Zur Verbesserung der Extraktion (Schmelzen der Fettphase) wird das Reagenzglas 5 Minuten im Wasserbad (5.4) auf 60 °C erwärmt. Danach wird das Reagenzglas sofort unter fließendem kaltem Wasser abgekühlt. Ein Teil des Extraktes wird durch ein Membranfilter (5.5) filtriert.
Das Filtrat ist am selben Tag zu chromatographieren.
- 6.1.2. Vorbereitung der Probe unter Zugabe des inneren Standards
1 g
0,1 g der Probe wird auf drei Dezimalstellen genau in ein Reagenzglas mit Schliffstopfen (5.3) eingewogen. Nach Zusatz von 0,50 ml Phosphorsäure (4.4), 0,50 ml der Lösung des inneren Standards (4.6) und 9 ml Ethanol (4.1) wird das verschlossene Reagenzglas kräftig geschüttelt. Zur Verbesserung der Extraktion (Schmelzen der Fettphase) wird das Reagenzglas 5 Minuten im Wasserbad (5.4) auf 60 °C erwärmt. Danach wird das Reagenzglas sofort unter fließendem kaltem Wasser abgekühlt. Ein Teil des Extraktes wird durch ein Mebranfilter (5.5) filtriert.
Das Filtrat ist am selben Tag zu chromatographieren.
- 6.2. Bedingungen für die Gaschromatographie
Folgende Betriebsbedingungen werden empfohlen:
Säule: | ||
Material | Edelstahl | |
Länge | 2 m | |
Innerer Durchmesser |
| |
Fester Träger | Chromosorb W AW, 100-120 msh | |
Stationäre Flüssigphase | 10% Polyethylenglycol 20 000 für Säuren (SPTM 1000 oder gleichwertiges Erzeugnis) und 1% H3PO4 | |
Temperatur: | ||
Injektor | 200 °C | |
Säule | 120 °C | |
Detektor | 200 °C | |
Trägergas: | ||
Stickstoff | ||
Volumenstrom: | 25 ml/min | |
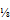
Säule: | ||
Material | Edelstahl | |
Länge | 2 m | |
Innerer Durchmesser |
| |
Fester Träger | Chromosorb W AW, 100-120 msh | |
Stationäre Flüssigphase | 10% Polyethylenglycol 20 000 für Säuren (SPTM 1000 oder gleichwertiges Erzeugnis) und 1% H3PO4 | |
Temperatur: | ||
Injektor | 200 °C | |
Säule | 120 °C | |
Detektor | 200 °C | |
Trägergas: | ||
Stickstoff | ||
Volumenstrom: | 25 ml/min | |
- 6.3. Chromatographie
- 6.3.1. Kalibrierung
In eine Reihe von 20-ml-Meßkolben werden 0,25, 0,50, 1,00, 2,00 bzw. 4,00 ml Propionsäurelösung (4.5) pipettiert. Nach Zusatz von 1,00 ml der Lösung des inneren Standards (4.6) mittels Pipette wird mit Ethanol (4.1) zur Marke aufgefüllt und gemischt. Diese Lösungen enthalten e mg/ml 2-Methylpropionsäure als inneren Standard (dh. 1 mg/ml, wenn e = 1,000) und p/4, p/2, p, 2p bzw. 4p mg/ml Propionsäure (dh. 0,25, 0,50, 1,00, 2,00 bzw. 4,00 mg/ml, wenn p =1,000).
1
Jede Lösung wird dreimal chromatographiert und der Mittelwert für das Peakflächenverhältnis berechnet.
Es wird eine Kalibrierkurve gezeichnet, indem das Verhältnis der Masse von Propionsäure zu 2-Methylpropionsäure als Abszisse und das Verhältnis der entsprechenden Peakfläche als Ordinate aufgetragen werden, oder es wird die Regressionsgerade berechnet.
- 6.3.2. Bestimmung
Jeweils 1
Die Chromatogramme der Probe- und der Standardlösungen werden verglichen. Falls das Chromatogramm der Probelösung einen Peak mit etwa der gleichen Retentionszeit wie 2-Methylpropionsäure aufweist, ist ein anderer innerer Standard zu verwenden. Wenn keine Interferenz beobachtet wird, wird 1
Die Probelösung wird dreimal chromatographiert und der Mittelwert für das Peakflächenverhältnis berechnet.
- 7. Berechnung
- 7.1. Aus der Kalibrierkurve (6.3.1) wird das Verhältnis der Masse (K) abgelesen bzw. aus der Gleichung für die Regressionsgerade berechnet, das dem gemäß 6.3.2 berechneten Peakflächenverhältnis entspricht.
- 7.2. Der Propionsäuregehalt in Prozent (x) der Probe wird unter Verwendung des ermittelten Masseverhältnisses nach folgender Formel berechnet:
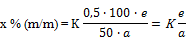
K = | nach 7.1 ermitteltes Masserverhältnis (Anm.: richtig: Masseverhältnis) |
a = | Einwaage der untersuchten Probe (6.1.2) in Gramm |
e = | Einwaage des inneren Standards (4.6) in Gramm |
Das Ergebnis wird auf eine Stelle nach dem Komma gerundet angegeben.
- 8. Wiederholbarkeit (1)
Bei einem Propionsäuregehalt von 2% (m/m) dürfen die Ergebnisse zweier an derselben Probe parallel durchgeführter Bestimmungen um nicht mehr als 0,12% (m/m) voneinander abweichen.
_________
(1) ISO 5725.
Zuletzt aktualisiert am
09.05.2017
Gesetzesnummer
10010899
Dokumentnummer
NOR12139954
alte Dokumentnummer
N8199658336J
Zusatzdokumente: image001, image002, image003, image004, image005, image006, image007, image008, image009, image010, image011, image012
Lizenziert vom RIS (ris.bka.gv.at - CC BY 4.0 DEED)